
Interested in the ethical conduct of human research? Want to know more about the Institutional Review Board (IRB) process? Become an IRB member! The Office of the Vice President for Research and ORI appreciates volunteers and is always on the lookout for interested and committed members. Contact Pam Stafford for more information (859-323-7399).
Institutional Review Board (IRB) Membership
The Office of Research Integrity (ORI) appreciates all of those who volunteer their time and expertise in IRB service!
REMINDER: IRB Member Confidentiality
Since IRB members and ORI staff have open access to all protocol information in E-IRB, required confidentiality measures must limit such access. Access is permissible only to protocols you are authorized/assigned for review or consultation purposes. This policy provides protection of intellectual property and proprietary information equivalent to our previous paper files. In order to remind IRB members of the scope of these protections, the member Confidentiality Agreement has been updated, parts of which now specifically reference E-IRB.
Please contact Pam Stafford via email or at 859-323-7399 if you have any questions.
REMINDER: IRB Member Conflict of Interest
In response to the Public Health Service updates for the investigator conflict of interest, the IRB member threshold has changed from $10,000 to $5,000. Detailed information is available in the IRB Member and Consultant Conflict of Interest SOP. The $5000 threshold is also reflected in the Conflict of Interest Statement that members sign at IRB orientation and annually thereafter.
Please remember that it is the responsibility of each voting member or alternate member of the IRB to disclose any conflict of interest when conducting any type of review and to excuse themselves from deliberations and voting if the review occurs at a convened meeting.
As the institution implements the new investigator COI policy and disclosure procedures, additional changes may be forthcoming. We will keep you informed of any additional changes impacting the IRB member COI.
Administrative Regulation (AR)
A new AR titled "Administration of Research Activities" will replace AR 7:4 (formerly III-4.0-6) "Human Research Subject Protection and Institutional Review Boards." COMING SOON!
Medical and Nonmedical IRB Meeting Dates & Membership
Checklists & Forms/SOPs
Checklists & Forms
UK IRB/ORI Standard Operating Procedures Pertaining to IRB Membership
IRB Member Guidance
Primary Reviewer Responsibilities
- Comparing the detailed grant application or industry protocol with the IRB application;
- Informing the full IRB of any discrepancies between the detailed protocol and the summary application materials;
- Determining whether the project involves a NIH multi-center clinical trial (e.g., cooperative group trial) and, if so, comparing the “Risks” and “Alternatives” section of the NIH-approved sample informed consent document with the UK proposed form to ensure that the NIH and UK sections of the consent are consistent;
- Ensuring that a Data and Safety Monitoring Plan (DSMP) exists IF research is greater than minimal risk or an NIH-funded or FDA-regulated clinical investigation;
- Reviewing the financial disclosure questions and alerting the IRB if a “yes” disclosure is made;
- Reviewing the other committee review/final approvals for consistency in human subjects protection measures;
- Checking the Signature Assurance sheet for appropriate signatures; and
- Conducting an in-depth review.
Vice Chair/Continuing Review (CR) Primary Reviewer Reference Guide [D77.0000]
Federal Requirements
- Office for Human Research Protection (OHRP) Guidance on IRB Continuing Review of Research, November 10, 2010
- Food and Drug Administration (FDA) Information Sheets: Continuing Review After Clinical Investigation Approval, February 2012
UK Standard Operating Procedures (SOPs)
- Initial Full Review SOP [C2.0100]
- Continuation and Annual Administrative Review SOP [C2.0250]
- Study Closure SOP [C4.0200]
UK Process
- CR Primary Reviewer Checklist Flowchart
- Current PI/SP Not Retrained
- Circumstances from Closure
- CR Screening/Review Responsibilities
- Process Outline for HIPAA Issues ID at Continuing Review
- Major/Minor Guidance
- Reportable IRB Determinants, Activities, and Definitions
- Convened IRB Requirements
Forms
IRB Reviewer Determinations (E-IRB)
Guidance on whether convened IRB request should be designated as minor (vote #2) or major (vote # 3 or 4) [D71.0000]
The DHHS Office of Human Research Protection policy states that contingent approval of revisions without subsequent review by the convened IRB (i.e., expedited review) is permitted only when the requested revisions are non-substantive (minor).
A vote of #2 at a convened meeting indicates that the IRB has given the individual chairing the meeting (and/or other IRB member with appropriate expertise or qualifications) the authority to approve minor revisions that do not involve substantive issues. The types of revisions that can be approved by expedited review must be directive (specific changes or revisions requested of the investigator to secure approval) or non-substantive (a change in which the judgment of the IRB reviewer, makes no substantial alteration in risks to subjects, selection of subjects, informed consent process, informed consent documentation, safety and monitoring or subjects’ privacy or data confidentiality. Examples of these are as follows:
- Changes in study research personnel;
- Adding a blood draw to a research study;
- Decreasing the amount of blood drawn or the frequency of blood drawn;
- Adding research site(s) to a research study;
- Adding a standardized test instrument to a research study;
- Changes to improve the clarity of statements or to correct typographical errors without altering the content or intent of the statement (e.g., minor changes in the consent form).
When the convened IRB requests substantive (major) clarifications or revisions (i.e. vote # 3 or 4) directly relevant to the regulatory determinations required by the IRB, or does not have the information needed to determine whether the regulatory criteria are met, the protocol revisions must be deferred to the convened IRB for review and approval and can not be reviewed and approved by expedited review procedure.
For example, review of requested revisions by the convened IRB should occur when there are changes that are substantive and non-directive, or there are questions, clarifications, or requests for information, such as:
- Clarify whether participants will be offered counseling services at the end of the study;
- Explain why participants less than 18 years of age will be allowed to participate;
- Provide additional information regarding study endpoints;
- Provide corrected informed consent documents (when incorrect consent documents have been submitted).
Review Outcome(s) for Full Review
An IRB member makes a motion, another member seconds the motion, and then the convened IRB votes for or against or abstains from one of the following five actions:
APPROVED (Vote for a #1): IRB approval - A vote for a #1 indicates that the IRB has concluded that the research and consent/assent forms meet the federal criteria for approval. IRB approval verifies that the IRB agrees with the assessment of the protocol and/or specific findings as described by the PI in the application. ORI staff will send the investigator an approval letter, according to the guidelines in the ORI Customer Service Standards, accompanied by an informed consent/assent document (if applicable) with the affixed "IRB Approval" validation stamp, which includes valid dates of IRB approval. If the IRB approves a HIPAA Waiver of Authorization Request, ORI staff will send a separate approval letter as well. (See the Mandated Reporting to External Agencies SOP [C4.0150].)
REVISIONS and/or ADDITIONAL INFORMATION REQUIRED (Vote for a #2): A vote of #2 indicates that the IRB has given the individual chairing the meeting the authority to approve the minor revisions. The IRB withholds approval pending submission of minor revisions/additional information. ORI staff will send the investigator a letter, according to the guidelines in the ORI Customer Service Standards, describing the revisions requested by the IRB.
The PI responds to the IRB’s suggested revisions in writing and sends the response to the ORI, which gives the response to the IRB Chair or member who chaired the meeting for further review. The Chair or designee may forward the responses to the entire IRB for additional review, request additional information, or approve.
TABLED (Vote for a #3): A vote of #3 indicates that the IRB withholds approval pending submission of major revisions/additional information. ORI staff will send the investigator a letter, according to the guidelines in the ORI Customer Service Standards. The letter lists the reasons for tabling and includes a description of the revisions or clarifications requested. For some studies, the IRB may appoint one or more members of the IRB to discuss the reasons with the investigator. If the vote is for a #3, ORI staff will schedule the PI’s response to the requested revisions for review by the full committee; the IRB does not require the PI to attend.
TABLED (Vote for a #4): If the vote is for a #4, the IRB follows the same procedure as for a vote of #3. In addition, ORI staff request that the PI attend the future IRB meeting at which the IRB reviews his/her response to requested revisions.
DISAPPROVED (Vote for a #5): If the vote is for a #5, ORI staff send the investigator a letter describing the reasons for disapproving the protocol. Disapproval of a protocol usually occurs when the IRB determines that the risk of the procedures outweighs any benefit to be gained or if the proposed research does not meet the federal criteria for IRB approval.
Research Risk Assessment Guidance
Digital Data Considerations
IRB Review of Medical Device Research [D110.0000]
The following question, definitions, and scenarios provide guidance for the evaluation of medical device research.
What is a Medical Device?
A medical device is an instrument, apparatus, implement, machine, contrivance, implant, in vitro reagent, or other similar or related article, including any component, part, or accessory, which is recognized in the official National Formulary, or the United States Pharmacopeia, or any supplement to them, intended for use in the diagnosis of disease or other conditions, or in the cure, mitigation, treatment, or prevention of disease, in man or other animals, or intended to affect the structure or any function of the body of man or other animals, and which does not achieve its primary intended purposes through chemical action within or on the body …. and which is not dependent upon being metabolized for the achievement of its primary intended purposes (Federal Food, Drug, and Cosmetic Act).
When do FDA regulations NOT apply?
FDA regulations would generally not apply to studies:
- using an FDA-approved device to test a physiologic principle where no data is collected about the device;
- using an FDA-approved device to address a research question, and no data is collected about the device; or
- using an FDA-approved device for clinical purposes (e.g., monitor a side effect, measure treatment progress);
as long as there is no intent to collect safety or effectiveness data or develop the device for marketing.
An example would be the use of an MRI to measure a clinical outcome in a study that has nothing to do with the MRI itself.
However, if the device used for one of these purposes is home-made by the principal investigator (PI) (e.g., a lever designed to raise the arm to measure flexibility), the informed consent should state that the device is not approved by the FDA.
When do the FDA-informed consent and IRB approval regulations apply?
FDA regulations apply when a study evaluates the safety or effectiveness of a medical device in subjects, healthy control subjects, or human specimens?
FDA Informed Consent and IRB (21 CFR 50, 56) regulations apply. The PI must include FDA language/references when developing the informed consent documents.
When is an Investigational Device Exemption (IDE) required?
FDA Investigational Device Exemptions (IDE) (21 CFR 812) regulations may apply for studies designed to:
- support marketing applications;
- collect safety and effectiveness information (e.g., for a new intended use of a legally marketed device); and
- sponsor-PI studies of an unapproved device or a new intended use of an approved device,
even if no marketing application is planned.
What is an Investigational Device Exemption (IDE)?
An IDE allows the investigational device to be used in a clinical study in order to collect safety and efficacy data required to support a marketing application. The term “exemption” in this case means exempt from laws prohibiting unapproved products from moving in interstate commerce.
What are the three regulatory categories for device studies described in the IDE regulations (21 CFR 812)?
Research that involves assessing the safety or effectiveness of a medical device must fit in ONE of the following categories:
- Studies exempt from IDE requirements (see scenarios 1 & 3 below);
- Significant Risk(SR)* device research with formal IDE submission to FDA (see scenario 2 below);
- Non-Significant Risk (NSR) device research, which, with IRB approval, is “considered” to have an approved IDE; sometimes referred to as an Abbreviated IDE (see scenarios 3 & 4 below).
*Significant risk device is an investigational device that: (1) is intended as an implant and presents a potential for serious risk to the health, safety, or welfare of a subject; (2) is for use in supporting or sustaining human life and represents a potential for serious risk to the health, safety, or welfare of a subject; (3) is for a use of substantial importance in diagnosing, curing, mitigating, or treating disease or otherwise preventing impairment of human health and presents a potential for serious risk to the health, safety, or welfare of a subject; or (4) otherwise presents a potential for serious risk to a subject. The IRB should evaluate the device as used in the study.
Figure 1: Diagram of regulatory categories for device studies.
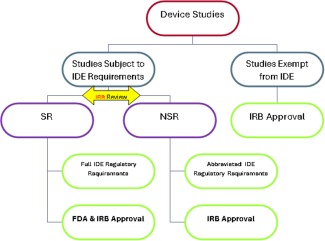
Who decides whether a device study is SR or NSR?
Sponsors are responsible for making the initial risk determination and presenting it to the IRB. Unless the FDA has already made a risk determination for the study, the IRB must review the sponsor's SR or NSR determination for every investigational medical device study reviewed and modify the determination if the IRB disagrees with the sponsor. If the FDA has already made the SR or NSR determination for the study, the agency's determination is final.
The following scenarios illustrate category determinations and procedures.
- The device section submitted in the IRB application indicates the device, as used in this study, is “Exempt” from IDE requirements.
- The PI designates the applicable category for IDE exemption on the IRB application. The IRB considers information provided by the PI, sponsor, or FDA that indicates that the device meets one of the exempt categories (21 CFR 812.2). If the IRB agrees that the study is exempt from IDE requirements, the IRB does not need to make the SR/NSR determination (see Figure 1) and may proceed to evaluate the study based on IRB approval criteria and informed consent regulations. However, if unsure, the IRB may request that the PI consult with the FDA to verify that the study is exempt from IDE requirements.
- NOTE: IDE-exempt studies are still subject to informed consent and IRB review regulations (21 CFR 50, 56); therefore, informed consent documents should still include a reference to FDA (e.g., FDA may view portions of your records). While not subject to routine inspection, IDE-exempt trials could still be inspected in response to a problem or issue with the device.
- Marketed Medical Device – Approved Indications
- The most common IDE exempt studies are those involving a marketed medical device in which the device is used or investigated in accordance with the indications in the cleared labeling. For these studies, the PI should provide, and the IRB should review, label information to compare the intended use in the protocol with the approved indications. Device approval indications may also be found by searching one of the FDA Device Approvals and Clearances databases.
- Studies not testing safety or effectiveness (e.g., consumer preference)
- Other examples of studies exempt from IDE requirements are consumer preference testing, testing of a device modification, or testing of a combination of two or more devices in commercial distribution if the testing does NOT collect safety or effectiveness data, or put subjects at additional risk.
- In Vitro Diagnostic (IVD) Devices
- In addition, diagnostic device studies (e.g., in vitro diagnostic studies) are exempt from the requirements under certain circumstances. The study is exempt as long as the sponsor complies with the requirements at 21 CFR 809.10(c) for labeling, and if the testing: (i) is noninvasive; (ii) does not require an invasive sampling procedure that presents significant risk; (iii) does not by design or intention introduce energy into a subject; and (iv) is not used as a diagnostic procedure without confirmation of the diagnosis by another, medically established diagnostic product or procedure. [21 CFR 812.2(c)(3)].
- Marketed Medical Device – Approved Indications
- The study is being conducted under a valid FDA-approved IDE.
- Office of Research Integrity (ORI) staff validate the IDE number, and no further IDE determination is required.
- The PI includes the IDE number in the IRB submission, and ORI staff use one of the following to validate the IDE number:
- Written communication from the sponsor;
- Written communication from FDA (required for PI held IDE;)
- Sponsor protocol imprinted with IDE #.
- The study is submitted with correspondence from the FDA indicating that the device is NSR or that the study is exempt from IDE requirements.
- The IRB does not have to make the SR or NSR determination as the FDA is considered the final arbitrator.
- The device, as used in the study, is NOT exempt, and the study does NOT have a valid IDE or FDA correspondence stating the device is NSR. THE CONVENED IRB MUST MAKE A SR/NSR DETERMINATION.
- The IRB makes the SR or NSR determination based on the proposed use of the device in the study. The SR/NSR determination is made by the full IRB at a convened meeting using information such as the sponsor’s risk designation and justification criteria, a description of the device, reports of prior investigations, the proposed investigational plan, and subject selection criteria.
- If the IRB determines the study is NSR, the IRB may approve the study using the standard approval criteria at 21 CFR 56.111. The study may begin without the submission of an IDE application to the FDA. The PI must still follow abbreviated regulatory requirements, including labeling, informed consent, monitoring, records, reports, and the prohibition on promotion. Progress and final reports are submitted only to the IRB.
- If the IRB disagrees with the sponsor’s NSR assessment and decides the study is SR, the IRB must tell the clinical PI, and where appropriate, the sponsor. (21 CFR 812.66)
SR device studies must have an IDE application approved by the FDA before they may proceed. The PI follows full regulatory requirements under the purview of both the FDA and the IRB. - NOTE: The device SR/NSR determination should not be confused with the “minimal risk” determination or “risk-benefit” assessment for the study in general.
- The IRB makes the SR or NSR determination based on the proposed use of the device in the study. The SR/NSR determination is made by the full IRB at a convened meeting using information such as the sponsor’s risk designation and justification criteria, a description of the device, reports of prior investigations, the proposed investigational plan, and subject selection criteria.
ORI staff document the decision of the IRB (both risk assessment and approval) in meeting minutes and in correspondence sent to the PI.
What if the FDA must be consulted when the IRB is unsure whether an IDE is needed?
The IRB, at its discretion, may contact or require that the PI contact the FDA for a determination.
Call 240-276-4040 with general questions about whether a product is subject to IDE regulations.
The FDA also recommends that the sponsor-PI contact the Center for Devices and Radiological Health (CDRH) if unsure about exemption from IDE requirements. To obtain a written risk determination from the FDA, submit correspondence labeled “Study Determination” in triplicate to the USFDA, CDRH, Document Mail Center – WO66-G609, 10903 New Hampshire Ave., Silver Spring, MD 20993-0002. CDRH Manufacturer’s Assistance may be reached at 800-638-2041, 301-796-7100, or via email.
Does device classification (Class I, II, III) factor into IRB determinations?
A sponsor’s detailed protocol may list a device as Class I, II, or III. FDA classifies devices based on the level of control necessary to assure safety and effectiveness of the device for marketing (not research). Controls range from general requirements such as labeling, not misbranding, and good manufacturing practices to special controls such as specific instructions for use or post-marketing surveillance requirements.
The classification is risk-based, so it is indicative of the type of submission required for the FDA to clear a device for marketing. Class II and Class III devices require the type of marketing route that most often involves clinical trials. Therefore, these are the types of devices seen in research for which the IRB is involved in the regulatory determinations addressed in the questions above.
Which regulatory device categories may be eligible for Expedited IRB review?
For a device study to be eligible for Expedited Review under Expedited Category 1, the device must present no more than minimal risk to the subject, and meet one of the criteria in Category 1b:
Expedited Category 1b- Research on medical devices for which:
- An IDE application (21 CFR 812) is not required*; or
- The medical device is cleared/approved for marketing, and the medical device is being used in accordance with its cleared/approved labeling**.
*Example: Study presents documentation from FDA indicating that an IDE application is not required, or the study meets all criteria to be an IDE-exempt in vitro diagnostic device 21 CFR 812.2(c)(3).
**An approved Device used in research according to its approved labeling is considered Exempt from IDE requirements - 21 CFR 812.2(c)(1 or 2). See scenario 4 above.
Note: Expedited Category 1 should not be used for studies that involve the use of a device only (no testing or data collected on or about the device), as FDA regulations do not apply. Expedited Category 4 may be considered for studies that use, but do not test, a device.
What else must the IRB consider in regard to PI responsibilities?
The IRB reviews the PI’s plan for management, control, and accountability of the investigational device. The ORI quality improvement program (QIP) resource website provides tools and sample standard operating procedures for device accountability. The ORI performs periodic QIP reviews of device studies to assess device control, access, and accountability.
What if the PI is also the sponsor of a device study?
The PI must indicate in the IRB submission that they are aware of their regulatory responsibilities in acting as both the PI and sponsor for an FDA-regulated investigation. The PI indicates if any responsibilities have been formally transferred to a contract research organization or other entity. Sponsor-PIs must complete the required Device Development for Sponsor-PIs’ Good Clinical Practice course available on CITI. ORI receives course completion notices directly from CITI.
What if the submission involves a Humanitarian Use Device (HUD)?
There are different requirements for HUDs depending on the device used in the study. See the Humanitarian Use Device SOP [C3.0200], IRB Summary - Humanitarian Use Devices, or FDA's Humanitarian Device Exemption (HDE) Regulation: Questions and Answers.
What if the submission involves a combination product?
A combination product comprises two or more regulated components (i.e., drug/device, biologic/device, drug/biologic) such as a prefilled insulin injector pen, transdermal patch, or metered dose inhaler. The FDA considers the whole combination when determining the need for an Investigational New Drug (IND) or IDE application. An application may be indicated if one part is new, one part increases the risk of an approved device/drug, or both drug and device are approved for different indications, and when two are used together, they present a new risk. The FDA Office of Combination Products website provides answers to frequently asked questions. hey may also be reached by email or phone (301-796-8930).
What if the submission involves a Compassionate Use or Treatment IDE?
Compassionate use allows access for patients who do not meet the eligibility criteria in a clinical investigation but for whom the treating physician believes the device may provide a benefit in treating and/or diagnosing a serious disease or condition. A treating physician or PI would contact the sponsor to request access to the device for an individual or a small group of patients. The sponsor submits an IDE supplement requesting approval of the compassionate use from the FDA. The IRB chair documents concurrence with the use, ensures the FDA has approved the use, and, after administration, receives and reviews reports of the use.
Treatment IDEs facilitate the availability of promising new devices to patients with life-threatening or serious diseases for whom no comparable or satisfactory alternative exists. Standard IDE regulations for conduct and IRB review apply to the Treatment IDE as data is collected on the device’s safety and effectiveness. A treating physician who uses a device under a Treatment IDE is responsible for meeting all applicable IDE responsibilities.
Sources:
- Information Sheet Guidance For IRBs, Clinical PIs, and Sponsors, Significant Risk and Nonsignificant Risk Medical Device Studies, January 2006
- Information Sheet Guidance For IRBs, Clinical PIs, and Sponsors Frequently Asked Questions About Medical Devices, January 2006
- Code of Federal Regulations, 21 CFR 812, Investigational Device Exemptions
- IDE Definitions and Acronyms
Summary of FDA Regulations on Investigational Device Exemptions and Exemption from IDE Requirements [D97.0000]
An investigational device exemption (IDE) allows the investigational device to be used in a clinical study in order to collect safety and effectiveness data required to support a Premarket Approval (PMA) application or a Premarket Notification 510(k) submission to the FDA.
Investigations covered under the IDE regulation are subject to differing levels of regulatory control depending on the level of risk. The IDE regulation distinguishes between significant risk (SR) and nonsignificant risk (NSR) device studies. Submit the device information and investigational plan to the IRB for concurrence with the sponsor’s SR/NSR determination.
The Investigational Device Exemptions (IDE) regulations describe the following types of device studies:
- Studies exempt from IDE requirements
- Studies subject to IDE requirements-
- SR device research with formal IDE submission to FDA
- NSR device research, which, with IRB approval, is “considered” to have an approved IDE (sometimes referred to as an Abbreviated IDE). In this case, the IRB acts as a surrogate for the FDA.
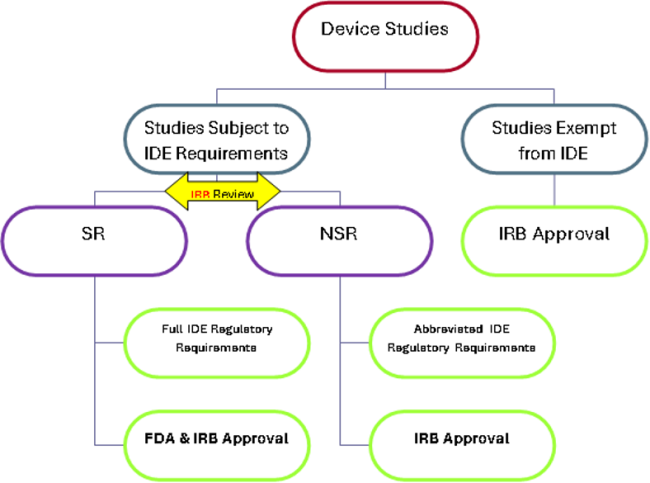
Is an IDE Needed?
Not typically needed:
- Practice of Medicine
- Basic physiological research
- Device used to:
- Test a physiological principle
- As a tool to address a research question
- No intent to:
- Collect safety and effectiveness data on the device
- Develop the device for marketing
- Device used to:
- IDE exempt studies (see below)
Yes would be indicated:
- Studies that support research or marketing applications
- Studies of new indications for an approved device
- Different age population
- New condition or disease
- Different areas of the body
- Change in indication (treatment, diagnosis, prevention)
- Significant design changes
- Study results will be submitted to the FDA
Studies Exempt from IDE
No IDE is required if the study meets one of the exemption categories in 21 CFR 812.2(c) that apply to human research. All criteria under each category must be true in order to meet the exemption category. IRB review and informed consent are still required.
Category 1-2
A clinical investigation with approved devices used in accordance with labeling. The device may have been approved for commercial distribution before May 28, 1976, or deemed substantially equivalent to a device commercially approved before May 28, 1976.
Category 3
A clinical investigation with in vitro diagnostic devices, if the sponsor complies with applicable requirements in 21 CFR 809.10(c), and if the testing:
- (i) is noninvasive;
- (ii) does not require an invasive sampling procedure that presents a significant risk;
- (iii) does not by design or intention introduce energy into a subject; and
- (iv) is not used as a diagnostic procedure without confirmation of the diagnosis by another, medically established diagnostic product or procedure. 21 CFR 812.2(c)(3).
Category 4
A clinical investigation with a marketed device undergoing consumer preference testing, testing of a modification, or testing of a combination of two or more devices in commercial distribution, unless testing is for determining safety and efficacy and/or puts subjects at risk.
*Categories 5 & 6 do not apply to human research.
Category 7
A clinical investigation of a custom device as defined in 21 CFR 812.2(b), unless the device is being used to determine safety or effectiveness for commercial distribution.
Submission and Regulatory Requirements for SR and NSR Device Studies
Clinical studies with SR devices must be approved by the FDA and by an Institutional Review Board (IRB) before the study can begin. Studies with NSR devices must be approved only by the IRB before the study can begin. Ongoing regulatory requirements apply to both.
Significant Risk Device Studies (standard IDE)
- Definition: A significant risk device means an investigational device that
- Is intended as an implant and presents a potential for serious risk to the health, safety, or welfare of a subject;
- Is purported or represented to be for a use in supporting or sustaining human life and presents a potential for serious risk to the health, safety, or welfare of a subject;
- Is for a use of substantial importance in diagnosing, curing, mitigating, or treating disease, or otherwise preventing impairment of human health, and presents a potential for serious risk to the health, safety, or welfare of a subject; or
- Otherwise presents a potential for serious risk to the health, safety, or welfare of a subject. (21 CFR 812.3(m))
- FDA Submission Requirements:
- Sponsors are responsible for making the initial risk determination and ensuring the investigator presents it to the IRB.
- Unless the FDA has already made a risk determination for the study, the IRB must review the sponsor's SR or NSR determination and modify the determination if the IRB disagrees with the sponsor.
- If the FDA has already made the risk determination, the IRB does not need to duplicate this effort.
- Sponsors of investigational SR device studies must have an IDE application approved by the FDA and IRB approval of the study before they may proceed.
- Ongoing Regulatory Requirements: SR device studies must follow all regulatory requirements in the IDE regulations in 21 CFR 812.
Nonsignificant Risk Device Studies (abbreviated IDE)
- Definition: An NSR device study is one that does not meet the definition for an SR device study.
- FDA Submission Requirements:
- Sponsors are responsible for making the initial risk determination and ensuring the investigator presents it to the IRB.
- Unless the FDA has already made a risk determination for the study, the IRB must review the sponsor's SR or NSR determination and modify the determination if the IRB disagrees with the sponsor. If the FDA has already made the risk determination, the IRB does not need to duplicate this effort.
- However, if the FDA has not made the risk determination or the IRB disagrees with the NSR determination made by a sponsor, then the IRB must notify the investigator and, where appropriate, the sponsor that the study involves a significant risk device.
- If the IRB agrees with the NSR determination and approves the study, the study is “considered” to have an approved application for IDE. No formal submission to the FDA is required. The IRB serves as an FDA surrogate for NSR studies.
- Ongoing Regulatory Requirements: NSR device studies must follow abbreviated regulatory requirements in 21 CFR 812.2(b).
Where questions still exist, sponsor-investigators should contact the appropriate FDA review division for guidance.
Contacts for the Center for Devices and Radiological Health
- 800-638-2041 or 301-796-7100
- IDE Inquiries: 301-796-5640
- Email CDRH
The FDA website outlines the FDA’s Procedure for responding to inquiries regarding the need for an IDE - Investigational Device Exemption (IDE)
Sources:
- Information Sheet Guidance For IRBs, Clinical PIs, and Sponsors, Significant Risk and Nonsignificant Risk Medical Device Studies, January 2006
- Information Sheet Guidance For IRBs, Clinical PIs, and Sponsors Frequently Asked Questions About Medical Devices, January 2006
- Code of Federal Regulations, 21 CFR 812, Investigational Device Exemptions
- PRIM&R webinar, Investigational Device Exemption Overview, Marian Serge, RN, Division of Bioresearch Monitoring, Office of Compliance, CDRH, FDA, 2009
Guide for IRB Review of Phase I Drug Trials [D136.0000]
Phase I trials are often conducted with healthy individuals to assess the safety and tolerability of a new drug or biologic. Trials involve a small group of participants in a controlled environment under tight timelines. Phase I trials may also be conducted in a patient population. Phase I oncology trials typically include cancer patients with no further treatment options.
Sponsors provide preclinical animal and toxicology data, previous clinical data if available, along with manufacturing information and proposed protocols to the FDA in an Investigational New Drug (IND) application. FDA’s approval of the IND is based on the assessment that the product is reasonably safe for use in initial, small-scale clinical studies.
Phase I trials assess the effects of the drug on the body (pharmacodynamics) and the effects of the body on the drug (pharmacokinetics) in terms of absorption, distribution, metabolism, and excretion (ADME). In addition, they may assess dose escalation, pharmacogenomics, food-drug or drug-drug interactions.
- Adequate Preclinical Data
- Ask for and consider preclinical results, including in vitro and animal studies. These have limited ability to indicate risk or predict effects in humans. Data from any prior use in humans should be presented to the IRB
- Investigational New Drug (IND)
- Confirm Phase 1 trials of drugs or biologics are being conducted under an FDA-approved Investigational New Drug (IND) submission
- Study Design
- If it includes a patient population, consider if placebo-controlled; justification for lack of crossover design
- Consideration of current standard care
- Assess Personnel Qualifications & Facility Emergency Care Provisions
- Emergency Readiness & Contingency Plans
- Subject Selection
- Exclusion of subjects thought to be “at-risk” for a particular adverse event (AE)
- Therapeutic misconception for patients with no other treatment options
- Dosing schedule
- Use of Investigational Drug Service (IDS)
- Max safe starting dose, dose increments, dose escalation – small dose increases between cohorts
- Slow infusion vs. bolus dose
- Use Sentinel Dosing Schedules – consider the amount of time between dosing to assess potential adverse events in one subject, before dosing a second subject
- Safety Monitoring:
- Assess plan for identification of AE or unanticipated problems
- Predictable and unpredictable toxicities
- Lab data adequate to assess all organ systems and organs likely to be affected by a drug
- Stopping Rules and thresholds for lab abnormalities
- When AE is delayed, repeated administration can lead to accumulated toxicity
- Report per sponsor and IRB Policy on Unanticipated Problems and Safety Reporting
- Monitoring should extend at least 30 days after study or through the half-life of a study drug
- Compensation:
- Remuneration amount based on time/effort/number of procedures; not on risks
- Schedule prorated – should be no financial penalty for withdrawal due to adverse event
- Consider the economic status of the population being recruited relative to the potential for undue influence
- Consent Process Considerations:
- Effective use of Key Information
- Identifies purpose (safety, proof of concept); clear that first trial in man
- Therapeutic Misconception for Phase I oncology trials
- Explanation of standard of care alternatives
- Clear description of treatment assignment, dosing ranges, multiple arms, etc.
- Potential risks associated with a drug, research procedures, and quality of life
- Unforeseeable risks
- Financial cost of an oncology trial
- Clear that compensation is prorated and not tied to withdrawal
- FDA typically will retain data collected to the point of subject withdrawal
- Not typically considered to be an “applicable trial” for registration on Clinicaltrials.gov
- Institutional Biosafety Committee (IBC)
- Review may be required for experimental immunotherapy.
Additional Resources
References
- FDA Safety Considerations in Phase 1 Trials, Sumathi Nambiar, MD, MPH, November 2012
- Study Design & IRB Review of Phase I Healthy Clinical Trials, October 2015, Quorum Review IRB Kurzrock, R., & Stewart, D. J. (2013). Compliance in Early-Phase Cancer Clinical Trials Research. The Oncologist, 18(3), 308–313.
- Protecting Phase I Subjects: IRB Considerations June 2013, Advarra IRB
- Oncology Studies from an IRB Perspective, May 2016, Advarra IRB
Summary of FDA Regulations on Exemption from IND Requirements [D46.0000]
In general, Investigational New Drug (IND) regulations (21CFR312) apply in human research studies that involve the use of a drug (as defined in the Food, Drug, and Cosmetic Act (FD&C Act)) in a clinical investigation (as defined in 21 CFR 312.3) unless otherwise exempt from IND requirements as described below. The following summary includes exemptions based on the IND Regulations, determinations from the 2013 FDA IND Exemption Guidance, and examples from the 2004 FDA Guidance on IND exemptions for cancer treatment studies.
Where questions still exist, sponsor-investigators are encouraged to contact the appropriate FDA review division for guidance.
- For drug studies, an inquiry concerning the application of the IND regulations should be directed to the Chief, Project Management Staff, in the appropriate CDER review division.
- For biologics, the inquiry should be directed to the applications division of the appropriate review Office.
Note: The determination of need for an IND does not depend on whether the intent of the clinical investigation is commercial or non-commercial. Also, the number of subjects to be enrolled or the clinical condition of the subjects has no bearing on whether the study is subject to the IND regulations. Unless a study meets one of the exemptions below, it is subject to IND regulations.
Exemption for Clinical Investigations involving a Lawfully Marketed Drug(s)
The clinical investigation of a drug product that is lawfully marketed in the United States is exempt from the requirements of an IND if all of the following apply:
- The investigation is not intended to be reported to the FDA as a well-controlled study in support of a new indication for use, nor is it intended to be used to support any other significant change in the labeling for the drug;
- If the drug that is undergoing investigation is lawfully marketed as a prescription drug product, the investigation is not intended to support a significant change in the advertising for the product;
- The investigation does not involve a route of administration or dosage level or use in a patient population or other factor that significantly increases the risks (or decreases the acceptability of the risks) associated with the use of the drug product;
- The investigation is conducted in compliance with the requirements for review by an IRB (21 CFR 56) and the requirements for informed consent (21 CFR 50); and
- The investigation is conducted in compliance with the requirements of 21 CFR 312.7 (Promotion and sale of investigational drugs).
How do you determine whether a planned study will be used to support a new indication or other significant labeling or advertising claim?
Whether a planned clinical investigation will be used to support a new indication, other significant labeling change, or advertising claim may not always be known or apparent at the outset of the investigation. Generally, it seems reasonable to infer that the intent of any well-controlled trial of a marketed drug sponsored by the manufacturer of the drug would be to influence labeling or promotion in some way. On the other hand, the sponsor-investigator of an investigator-initiated study in an academic setting (a study designed and initiated by the investigator independent of the manufacturer) probably does not intend that his or her study of a marketed drug influence labeling or promotion, even if the sponsor-investigator is receiving some limited support from the drug’s manufacturer. However, certain investigator-initiated research has the potential to influence labeling or promotion, notwithstanding the investigator’s intent (e.g., a controlled trial with an endpoint representing improvement of a serious disease). Similarly, certain studies of effectiveness conducted by government agencies (e.g., National Institutes of Health, Veterans Administration) have the potential to influence labeling. FDA strongly encourages IND submissions for these types of studies so that the Agency can have an opportunity to provide advice on study design.
How do you determine whether changes to a lawfully marketed dosage form increase risk?
FDA does not require that the exact same dosage, population, or form described in approved labeling in order to meet the exemption category, but permits changes that do not increase the risks above those presented by the use of the product according to approved labeling.
Investigators are advised to carefully consider the risk implications of any conditions of use that deviate from those described in approved labeling, particularly in regard to route of administration, dose, and patient population.
- Route of Administration: A change in the route of administration can introduce a significant new risk. For example, there could be a significant increase in risk if a marketed drug for oral administration is converted to a dosage form that is to be administered by injection, intravenous, intrathecal, or inhalation route. These other routes of administration introduce concerns with sterility, pyrogenicity, hypersensitivity (e.g., airway reactivity), variations in metabolism, and other issues not present with oral administration.
- Dose: Increases in dose, frequency, or duration of administration, compared to labeled dosing regimens, can significantly increase the risk in a study using a marketed drug. It is possible that a decrease in dose could also significantly increase risk. For example, administering a low dose of a pure polysaccharide vaccine to study subjects can induce hypo-immunologic or non-immunologic responses in the subjects and can also induce tolerance to the vaccine, thus making subjects at risk for the infectious disease the vaccine is intended to prevent. The significance of changes in dose (in particular, increases in dose) can vary across therapeutic areas. For example, the cancer treatment guidance provides some latitude for conducting studies of high-dose cancer treatments without an IND because of oncologists’ familiarity with the implications of high-dose regimens, generally.
- Population: The acceptability of known and unknown risks can vary considerably across different treatment populations (see 21 CFR 312.2(b)(1)(iii)). For example, a drug with significant toxicity can be approved for use in a population with life-threatening or severely debilitating disease because the risk of toxicity is acceptable in that population. Use of that drug in a clinical investigation in a population that is not so ill (e.g., to evaluate the drug for prevention of disease or symptomatic relief), however, would present a different risk-benefit situation in which the risks would likely not be acceptable. When the acceptability of the risk is significantly decreased, the study would have to be conducted under an IND as required under 21 CFR 312.
Exemption of Clinical Investigations involving In-Vitro Diagnostics
A clinical investigation of an in vitro diagnostic biological product is exempt from the requirements of an IND if all of the following apply:
- The in vitro diagnostic biological product involving one or more of the following:
- Blood grouping serum.
- Reagent red blood cells.
- Anti-human globulin.
- The diagnostic product is intended to be used in a diagnostic procedure that confirms the diagnosis made by another, medically established, diagnostic product or procedure.
- The diagnostic product is shipped in compliance with 21 CFR 312.160.
Exemption for a Clinical Investigation involving a Placebo
A clinical investigation involving the use of a placebo is IND-exempt if the investigation does not otherwise require submission of an IND. Note: additional requirements apply for research in children/minors based on the FDA Subpart D final rule.
Other Products
See 2013 FDA IND Exemption Guidance for additional FAQs regarding the need for an IND, including dietary supplements, live organisms, radioactive drugs, bioavailability or bioequivalent studies, radiolabeled peptides, positron emission tomography (PET) drugs, attenuated microorganisms, radioisotopes, and others.
Cancer Treatment Determinations
Below is additional guidance for when studies of lawfully marketed drugs or biological products, for the treatment of cancer, are exempt from the requirement of an IND application, and examples of when they are not.
When does an IND application need to be submitted for studies of marketed drugs for treating cancer?
(excerpt from 2004 FDA Guidance: IND Exemptions for Studies of Lawfully Marketed Drug or Biological Products for the Treatment of Cancer)
When determining if an IND needs to be submitted to study marketed drugs for treating cancer, investigators must apply the exemption criteria listed in 21 CFR 312.2(b)(1) in light of the discussion in this guidance. Planned studies may be considered exempt from the requirements of an IND if the studies involve a new use, dosage, schedule, route of administration, or new combination of marketed cancer products in a patient population with cancer, and the following conditions apply:
- The studies are not intended to support FDA approval of a new indication or a significant change in the product labeling;
- The studies are not intended to support a significant change in the advertising for the product;
- Investigators and their IRBs determine that, based on the scientific literature and generally known clinical experience, there is no significant increase in the risk associated with the use of the drug product;
- The studies are to be conducted in compliance with IRB and informed consent regulations, pursuant to 21 CFR 50 and 21 CFR 56;
- The studies will not be used to promote unapproved indications, in compliance with 21 CFR 312.7.
EXAMPLES OF STUDIES
Refer to the 2004 FDA Guidance: IND Exemptions for Studies of Lawfully Marketed Drug or Biological Products for the Treatment of Cancer for examples of studies that are being provided to illustrate the FDA’s current thinking on the types of studies that the FDA considers to be exempt from IND regulation based on a risk assessment.
IRB Member Training Resources
Annual Update for Alternate IRB Members
Annual Update for Alternate IRB Members Video
Latest developments, reminders, and insights for ethical research oversight.
Artificial Intelligence/Machine Learning in Human Research
For questions on whether software using AI or Machine Learning is a Medical Device, see the FDA Guidance below and/or contact Belinda Smith or Sam Bell with questions.
Artificial Intelligence/Machine Learning Human Research
FDA Final Clinical Decision Support (CDS) Software Guidance
Determine if Your Clinical Decision Support (CDS) Software is a Medical Device
Expedited Review Fundamentals for IRB Members
E-IRB Training for IRB Members
At your convenience, review the ABCs of E-IRB video tutorial series and the video tutorials for IRB Members in the E-IRB Video Tutorial Library to learn E-IRB Basics, navigation, and procedures for Primary Reviewers, and for Required Reviewers and Other Reviewers.
If, after reviewing the videos, you would like live online E-IRB training, please submit a request to E-IRB Support.
Informed Consent Waivers
Health Disparity Training for IRB Members
ORI has packaged the presentation by Dr. Lovoria Williams and Dr. Nancy Schoenberg, presented at the September 2020 IRB in-service, into an online training module hosted on myUK Learning. IRB members who were unable to attend the virtual in-service may request course enrollment to watch a video of the presentation. The course also includes an optional four-question quiz, which qualifies as Human Subject Protection (HSP) refresher training.
You will need to request course enrollment.
After submitting the request, you will receive an email confirming the course has been added to your learning plan on myUK Learning. After watching the recorded presentations and answering 3 of 4 questions correctly, you may download a completion certificate. The quiz may be repeated. ORI receives monthly completion notices and will update E-IRB training records monthly or upon request if you need your record updated sooner.
Email the HSP Training Support Team to request course enrollment or with any questions.
IRB Member News
COMING SOON!
"New IRB Member" Orientation Training Resources
- IRB Member Orientation Online Course: supplemental course on ethical foundations, IRB basics, and member responsibilities
- Collaborative Institutional Training Initiative (CITI) New IRB Member Training
- Log in to CITI through the Single Sign-On page using your Link Blue ID and current Link Blue password
- Scroll to the bottom of the page, under " Learner Tools for University of Kentucky", click "Add a Course"
- Choose Human Subject Protection, then click Next
- Choose Institutional Review Board (IRB) Members, then click Next
- Choose the applicable training:
- IRB Chair Training - Choose if you are a Chair or Vice Chair of an IRB
- IRB Member Training - Choose if you are a Regular Member or an Alternate Member of an IRB
- IRB Community Member Training - Choose if you are a Community Member of an IRB
- This adds the course to your Active Courses on the University of Kentucky Courses page
- Click Review Course
- Complete any quizzes (quizzes may be repeated)
- Log in to CITI through the Single Sign-On page using your Link Blue ID and current Link Blue password
Community Member Refresher Human Subject Protection Training
External Training Resources
Nuts & Bolts of Being an IRB Member
As federally mandated, the IRB is comprised of assorted members with varying backgrounds to promote complete and adequate review of research activities. IRB members learn about a variety of interesting research studies, the ethical issues surrounding human participation, and the mechanisms in place to protect human subjects. If you've ever wondered about the process or criteria for serving as a volunteer on the IRB, the following summary provides a snapshot of what is involved.
Eligibility:
IRB members are officially appointed by the Vice President for Research. Non-tenure track faculty, or tenure track Associate Professors or higher, are eligible for consideration as an IRB member or IRB alternate (this stipulation is the Vice President for Research’s (VPR) to help ensure a commitment to the IRB does not prevent someone on a tenure track from getting tenure). Individuals need to be willing to invest the time involved in performing IRB member/alternate responsibilities.
Training:
The Office of Research Integrity (ORI) provides initial IRB member orientation and assigns an experienced IRB member to serve as a mentor. Ongoing continuing education is provided through various venues, including quarterly IRB in-service sessions, topic-related educational materials attached to protocols, and education updates at regular IRB meetings.
Duration of Term:
A typical appointment term is three years.
Regular IRB Membership
For regular members, an investment of time involves a commitment to regularly attend convened meetings and periodically serve as an Exemption or Expedited reviewer. Candidates must also acquire approval from their department chair or the individual to whom they report.
Meeting Schedule:
Normally, regular IRB members have one meeting every three weeks on their committee's assigned weekday. Medical IRB meetings are currently held on either Monday, Tuesday, Wednesday, or Thursday and average about 1.5 hrs each. Nonmedical IRB meetings are currently held on Fridays and average about 2 hrs each. Periodically, a regular member of an IRB may be asked to serve as an alternate for any comparably qualified member on another IRB (e.g., none of the alternate IRB members have equivalent expertise or are available to substitute, so ORI needs to seek equivalent expertise from another committee).
Workload:
Current members report varying times involved in preparation for the meetings – anywhere from one hour to five hours. The time involved depends on the complexity of the study, the existence of regulatory issues, and the organization of the materials. Trained ORI staff do their best to ensure appropriate materials have been included with the protocol to help reviewers address applicable regulatory criteria. Materials for review are disseminated to members approximately ten days before a meeting.
Alternate IRB Membership
Schedule:
Should a regular member be unavailable for an IRB meeting, an IRB alternate who has equivalent expertise with the regular member may be called upon to review protocol materials and attend the IRB meeting in that regular member's place. There is no pre-determined schedule for this, and sometimes ORI only finds out a week or so in advance of the meeting that a substitute is needed.
Workload:
The time commitment for an alternate varies considerably, depending on the demand for the specific area of expertise and the availability of the IRB member with whom the alternate is paired. It's possible an alternate could go an extended period without being asked to conduct a review. However, IRB alternates may also serve on a monthly rotation as an Expedited Reviewer and/or Exemption Reviewer. Additional training is provided to enable members to become proficient in these streamlined review mechanisms. The IRB and ORI have Standard Operating Procedures (SOPs) describing policies and procedures for IRB reviewers in more detail.
For more information about volunteering to serve on an IRB, contact Pam Stafford.
Community IRB Membership (Unaffiliated and/or Nonscientist)
Federal regulations state that IRBs must be varied and must include three kinds of members: scientists, nonscientists, and people not affiliated with the institution.
The nonscientist is vital – research review cannot proceed without a nonscientist present. This member’s role is to ensure that research materials are readable and that participants will understand what they are volunteering for. This committee member must be:
- A nonscientist (training, background, and occupation are in nonscientific fields);
- Able to prepare for and attend most committee meetings; meetings are set for a particular weekday and are held about 3-4 weeks apart;
- Able to attend occasional education sessions; and
- Comfortable with basic computer operations.
The unaffiliated member brings a unique perspective to the table. This member’s role is to represent the public’s concerns – to serve the public community’s interests rather than the university’s. This committee member must be:
- Not affiliated with the University of Kentucky, either directly or through an immediate family member;
- Able to prepare for and attend most committee meetings; meetings are set for a particular weekday and are held about 3-4 weeks apart;
- Able to attend occasional education sessions; and
- Comfortable with basic computer operations.
IRB Membership Recruitment Brochures
For a downloadable brochure PDF, contact ORI.
Jump to: Medical IRB | Nonmedical IRB | Nonscientist Member | Unaffiliated Member
Medical IRB Member

Nonmedical IRB Member

Nonscientist IRB Member

Unaffiliated IRB Member

Concerns, Suggestions, or Questions?
Concerns regarding attempts of undue influence on individuals responsible for the oversight of human subjects research should be reported to the ORI Executive Director, who notifies the Vice President for Research (VPR). The VPR, in consultation with the ORI Executive Director, determines the appropriate response to unduly influence or undermine the mission of the IRB. The ORI Executive Director may be contacted by phone or email.